


推荐产品





公司新闻/正文
【深度】中检院发文:全面复盘10款CAR-T注册检验,这些“雷区”千万别踩!
172 人阅读发布时间:2026-02-06 11:34
近日,《中国细胞生物学学报》发表了一篇来自中检院发表的文章《CAR-T细胞治疗产品上市前注册检验的特殊要求及关注点》,该文章系统梳理了目前已完成注册检验的10款CAR-T产品(含8款已获批,2款NDA阶段)的实战经验。本文我们将为您提炼其中的核心干货,探讨监管视角下CAR-T注册检验的难点与破局之道。
传统大分子药物 vs. CAR-T:被放大的“时空困境”
与传统大分子药物(如单抗)成熟的生产模式不同,CAR-T产品的注册检验面临着先天性的基因限制。专家在文中明确指出了二者的显著差异:
- 大分子药物(如单克隆抗体):单批次产量大(满足成千上万患者)、稳定性好、有效期长(2-3年)。
- CAR-T细胞治疗产品:个体化生产、产量极低(无法按常规取样原则取样)、部分新鲜制剂有效期极短。
这就造成了注册检验的挑战:检验时限与样本量的博弈。 尤其在第一轮检验出现异常需复试时,极易因“没有备样”或“备样过期”导致审评延误。
官方破局方案:如何合理设置取样点与样本量?
中检院专家给出了极具指导意义的实操建议:
1. 过程样 vs. 终产品:以最适样本为原则
支原体、牛血清白蛋白及抗生素残留等项目可采用过程控制样本
外观、细胞活率、表型及活性等检测项目则可采用终产品样本
2. 健康供者样:患者样本不足时的合规备选
若患者来源的终产品样本不可行,可采用至少3个不同健康供者来源的、经商业规模生产的样本进行检验,但需充分评估其代表性风险。
3. 样本包装与复试的“底线思维”
为满足复试要求,原则上应提供至少4个独立包装的样品。
若样本量实在无法满足,申请人需提交 “不符合规定时不进行复试”的书面声明,并优先保证安全性项目的检测。
避坑指南:175条复核意见暴露了哪些“雷区”?
文章首次披露了对这10个CAR-T品种注册检验中提出的175条复核意见。数据显示,问题主要集中在以下三个方面,值得所有CGT企业警惕:
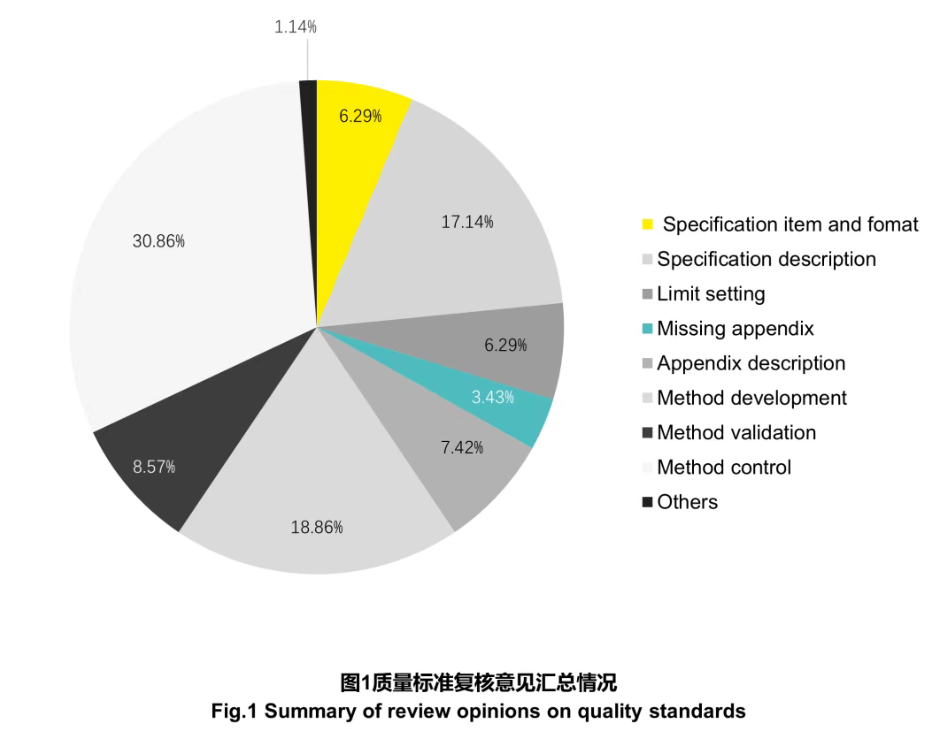
雷区一:质量标准“形似而神不至”
许多企业提交的质量标准在形式上看似完整,实则缺乏科学支撑。具体表现为:
- 表述不规范:如将“CAR阳性率”误称为“感染效率”,或将本应属于过程控制(IPC)的RCL检测错误列入成品放行标准。
- 限度脱离实际:尤其在生物学活性和残留物等关键指标上,未能基于验证批数据及临床反馈进行合理论证,导致限度设定过宽或过严。
雷区二:方法开发“有术而无验”
检测方法的建立常常止步于“可用”,而未追求“可靠”。
- 模型未验证:最典型的是直接套用商品化试剂盒的四参数模型,却未验证其对本品基质的适用性。
- 判定逻辑缺陷:如CAR基因鉴定中,仅要求扩增条带大小或仅要求测序,“二者缺一”导致无法形成确证产物序列的完整证据链。
雷区三:方法控制“有测而无控”
这是复核意见中占比最高(30.86%)的环节,意味着检测过程缺乏“纠错机制”。
- 参照物失效:未设质控品,或质控品浓度过高、缺乏代表性(如将标准曲线浓度点当作质控品)。
- 有效性标准空白: 缺少“参比品结果需在标定范围内”的系统适用性要求,以及复孔RSD等精密度要求。
- 必要对照缺失: 如效力评价中缺少阴性靶细胞对照,导致结果特异性无法判断。
核心问题关键点分析:以上雷区共同指向一个根本性短板:质量体系中的“质量控制思维”未能深度贯穿于从方法开发到标准制定的全过程。企业往往更关注“做出数据”,而忽视了如何通过科学的参比系统、内置质控和完整的判定逻辑来持续证明“数据的可靠性与可比性”。
延伸思考:无菌检测的破局思路
在文章中,专家反复强调了“时效性”与“样本量”。而在所有放行检验中,无菌检查无疑是最大的痛点——传统药典法需要消耗大量样本,且需等待14天。这对于有效期仅几十小时的新鲜CAR-T制剂来说,是绝对的商业化阻碍。
事实上,不仅是中国,全球监管机构都在加速推动无菌快检的替代。美国药典USP <1071> 就专门针对短货架期产品提出了Rapid Microbial Tests的指导原则,鼓励使用如包括核酸扩增法在内的快检技术,以减少样本消耗并将检测时间缩短至几天甚至数小时。
行业实践:让合规与效率并肩同行
看完专家的梳理我们不难发现,在保证安全的前提下,最大限度节约样本、缩短检验周期,是监管与企业的共同诉求。
作为CGT领域无菌快速检测的先行者,赛多利斯致力于提供符合全球药典标准的无菌快检方案:
Microsart® ATMP 无菌放行检测试剂盒专为快速检测细胞培养物、细胞培养物衍生生物制剂及 ATMPs 中的总细菌和真菌设计。该技术基于实时聚合酶链反应 (qPCR)。此包装版本包含 10 个患者样品放行检测所需的全套试剂。DNA 提取试剂与 PCR 试剂按单个待检样品独立包装,可有效避免交叉污染,确保操作便捷性最大化。该试剂盒对18 种细菌和7 种真菌(包括6种USP 和EP 要求的菌株)的检测灵敏度(5-99 CFU/ml)进行了验证,同时与基于药典的培养法进行了检测等效性的对比。(注:本检测不可替代依据 USP<71> 或 EP 2.6.1 开展的经典无菌检测。)
- 快速准确:仅需3小时即可获得检测结果,确保病人用药安全
- 减少移液操作:体系中包含内控,有效排除假阴性结果
- 符合国际标准:依据 EP 5.1.6 和 USP <1223> 标准完成灵敏度、特异性及稳健性验证
在CGT商业化的冲刺道路上,一个可靠、高效、合规的快速检测方案,不仅是提升效率的工具,更是保障患者可及性与产品成功上市的战略支点。我们愿以创新技术,与您一同跨越检验关卡,加速治愈的到来。